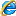因蛋白聚糖降解酶先天性缺陷所引起的蛋白聚糖分解代谢障碍,将导致产生各种类型的粘多糖沉积症(mucopo-lysaccharidoses)。其特征是过多的寡聚糖堆积与排泄。各型粘多糖沉积症的代谢基础相似,但遗传类型和临床表现各不相同。
【诊断】
粘多糖沉积症的酶缺陷
注:MPS-粘多糖沉积症DS-磷酸皮肤素
HS-硫酸乙酰肝素CD-硫酸软骨素
KS-硫酸角质素
遗传性粘多糖沉积症约占出生婴儿的1/30000。
【临床表现】
1、粘多糖贮积症I型(mucopolysaccharidosis)
又称Hurler综合征、承雷病。AR。为溶酶体a-L-艾杜糖醛酸酶缺乏,发病率1/10万新生儿。主征;进行性发育迟缓,体矮,智力低下,舟状头,颈短,面容粗陋,有角膜混浊,关节僵硬,活动受限,驼背。其他:视网膜色素沉着,耳聋,胸廊畸形,肝脾肿大,腹大,心脏瓣膜缺损和动脉硬化等。粘多糖筛查阳性,根据酶活性测定可检出携带者。预后差,多于儿童期死亡。
2、粘多糖贮积症II型(mucopolysaccharidosis)
又称Hunter综合征。XR,无女性患者,发病率为6/10万。为艾杜糖醛酸硫酸酯酶缺乏。主征:与I型相似,但无角膜混浊,背不驼。其他:多毛,进行性耳聋;肌肉痉挛,行为莽撞。10岁后2/3患者有惊厥发作。生化诊断如I型,主要根据临床表现相鉴别。重型者多在青春期前死亡。
【辅助检查】
粘多糖沉积病是由于细胞溶酶体酸性水解酶先天性缺陷所致。根据病因目前本病可分为八大类型,我国已报告200从余例,主要表现为严重的骨骼畸型、肿脾肿大,智力障碍以及其它畸形。粘多糖沉积病产前诊断以测定培养羊水细胞内特异的酶活力最为可靠,但实验要求高,一般实验室难开展。两种较简单的实用的方法是甲苯胺蓝定性及糖醛酸法半定量测定。
1.甲苯胺蓝定性:方法同尿液粘多糖检查。妊娠早期羊水正常者可呈阳性,妊娠中后期为阴性如阳性提示胎儿患粘多糖沉积病。
2.糖醛酸半定量测定:羊水中酸性粘多糖与四硼酸钠硫酸溶液反应生成糖醛酸,以每毫无肌酐中糖醛酸的量反映酸性糖多糖的多少。随着妊娠的进展,糖醛酸含量逐渐减少,妊娠16-20周的参考值为3.3~7.0mg/mgCr,如高于此值应考虑有粘多糖沉积肥病。本法除Morguio综合征外对其它类型的粘多糖沉积病均有诊断意义。
3.常染色体隐性遗传病(autosomal recessive disorder):致病基因在常染色体上,基因性状是隐性的,即只有纯合子时才显示病状。此种遗传病父母双方均为致病基因携带者,故多见于近亲婚配者的子女。子代有1/4的概率患病,子女患病概率均等。许多遗传代谢异常的疾病,属常染色体隐性遗传病。按照“一个基因、一个酶”(one gene one enzyme)或“一个顺反子、一个多肽”(one cistron one polypeptide)的概念,这些遗传代谢病的酶或蛋白分子的异常,来自各自编码基因的异常。常见的常染色体隐性遗传病有溶酶体贮积症,如糖原贮积症、脂质贮积症、粘多糖贮积症;合成酶的缺陷如血γ球蛋白缺乏症、白化病;苯丙酮尿症、肝豆状核变性(Wilson病)及半乳糖血症等。